cPEPmatch Tutorial
cPEPmatch is a computational tool designed to help you rationally design cyclic peptides that can potentially bind to specific regions of protein-protein and protein-ligand complexes. In this tutorial, we will walk you through the process of using cPEPmatch and provide an example for better understanding.
Step 1: Identify Your Target Complex
* Download PDB Structure: Start by identifying the protein-protein or protein-ligand complex you want to target. Download the crystal structure of your chosen complex from the Protein Data Bank (PDB).
S100P Example: For this tutorial, we'll use the S100P - V Domain of the receptor for advanced glycation end products (RAGE) complex with PDB ID: 2MJW. S100P is a calcium-binding protein that interacts with the RAGE receptor, and disrupting this interaction has therapeutic potential for various diseases.
Step 2: Inspect and Prepare the PDB File
* Inspect the PDB: Take a close look at the downloaded PDB file. If necessary, perform any cleanup, such as removing unnecessary chains, renumbering residues, and ensuring the correct PDB format. Ensure there are no missing residues in the binding site.
Tip: If there are missing residues outside the binding site, you can use tools like Modeller to fill them in for later simulations.
Step 3: Select cPEPmatch Parameters
* Cutoff: This parameter defines the distance between the receptor and the interface. Residues within this cutoff distance will be considered part of the interface. (Default: 6 Angstroms)
* Threshold: Set the fit-RMSD maximum for a match to fit into the targeted protein interface residues. It determines how well the cyclic peptide backbone should fit those of the matched protein residues. (Default: 0.7 Angstroms - Recommended: 0.3-1.5)
* Motif Size: This is the length of the motif residues to target. (Default: 5 amino acids, Recommended: Adjust based on the nature of your binding site—lower for pocket-like or larger for flat interfaces. Lower limit is 4, upper limit is 7)
* (Optional) Residue Numbers: Specify specific residues if you know the binding hot spots. Hot spots are residues contributing the most to binding free energy. Selecting this option allows non-consecutive motif searches. You can use experimentally known hot spots or calculate them computationally. When hot spot residues are selected, the cutoff is ignored.
Note:
* Hot spot searches may take more time.
* A higher threshold is recommended to accommodate non-consecutive residues - but no higher than 2.
* Ensure your hot spot count is at least as large as the motif size.
S100P Example: In our case, we identified hot spots for chain A binding to chain B as residues '28 32 34 37 41 43 49 78 90 92'. We set threshold=2 and motif=5. Refer to the figure below. On the left, you'll see an example of Chain A (in magenta) and Chain B (in yellow) at the binding interface, with the hotspots highlighted in red licorice. On the right, an example of the submission form is displayed
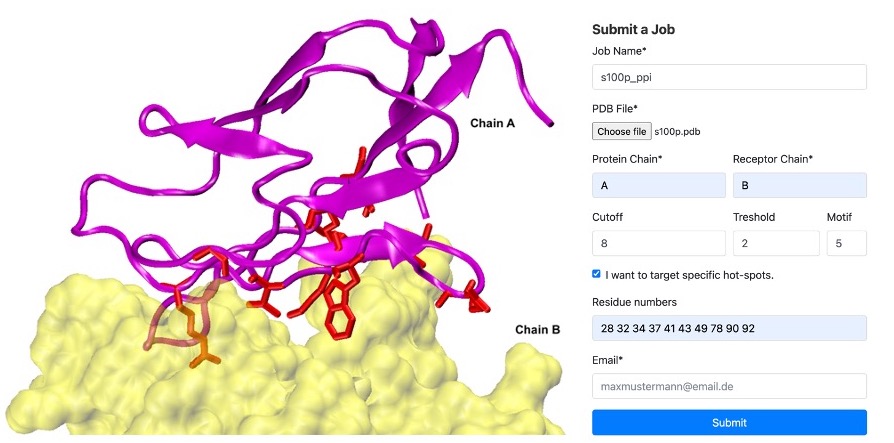
Step 4: Run cPEPmatch
* Output: Submit your job, and after some time, you will receive an email with a link to download a zip file containing your results. Be sure to check your spam folder.
The zip file contains the following files:
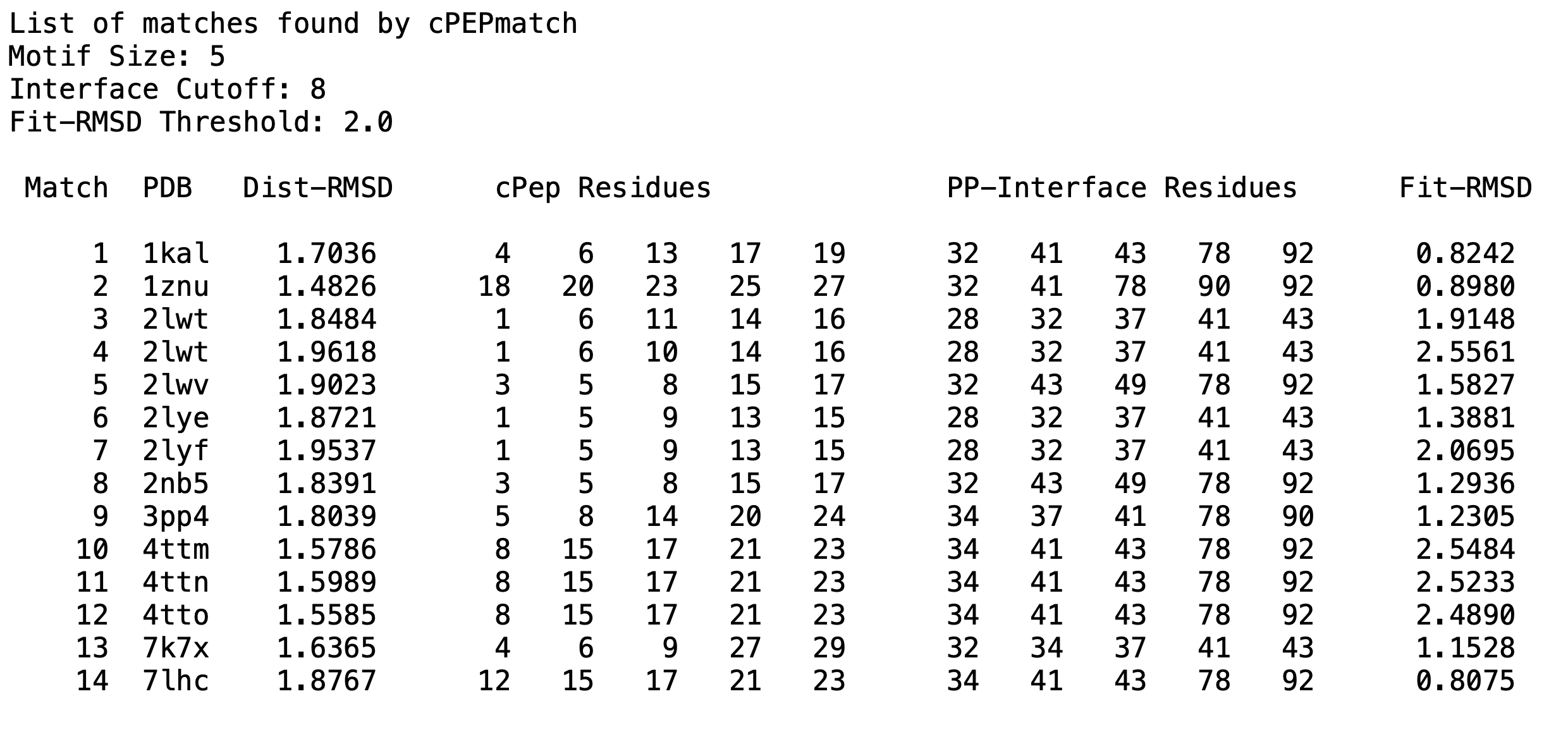
* match_list.txt file listing all matches, their fit values, and which cyclic peptide residues matched with the protein.
* match#_PDB.pdb files: all your matches' cyclic peptides, which have been superimposed, mutated and refined using Modeller and MD to fit the matched residues. Note: If the peptide contains non-standard residues, mutations will not be executed, and the output file will have "_non-mutated" in the name. Tip: You can perform the mutations using Amber Tools or Modeller.
* interface.pdb file: this is a PDB file with the residues that were selected as the interface>
* Log files.
S100P Example: cPEPmatch identified 14 matches using the provided example parameters. Below is a representation of what the output file, named 'match_list.txt', contains:
Step 5: Post cPEPmatch Evaluation
* Visual Inspection: Visually inspect your matches and identify the best-sterically fitting ones.
* MD Simulations and HREMD: Evaluate your matches using Molecular Dynamics (MD) simulations or Hamiltonian Replica Exchange Molecular Dynamics (HREMD). Calculate the binding free energy to select the best matches as lead structures for further binding optimization.
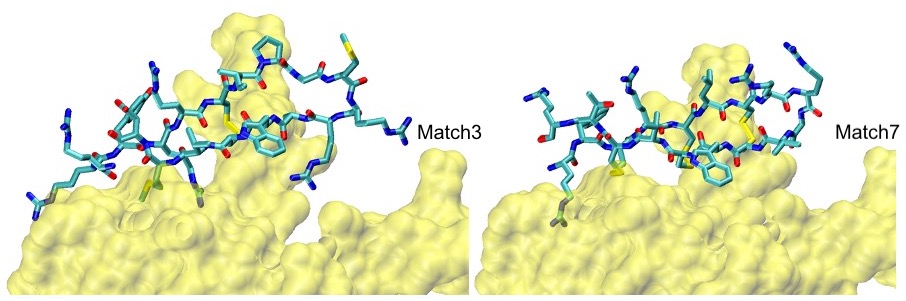
S100P Example: Our top two scoring matches, confirmed MD and HREMD simulations, are shown below:
Download the S100P Example output zip file here.
Congratulations! You've completed your cPEPmatch and hopefully have promising lead structures for further optimization. If you didn't find suitable binders, consider experimenting with different matching parameters.
Enjoy the peptide design process and good luck with your research!